Typical reductions in A1C values - Incretin Mimetics:
Overview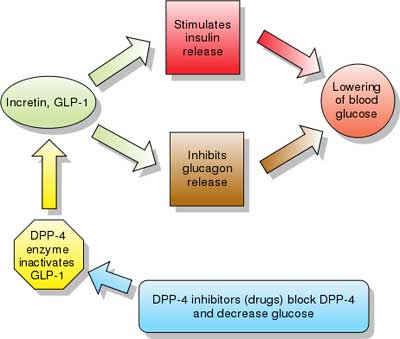 |
| Drug UPDATES: TRULICITY ™ (dulaglutide) injection, for subcutaneous use [Drug information / PDF] Dosing: Click (+) next to Dosage and Administration section (drug info link) Initial U.S. Approval: 2014 Mechanism of Action: TRULICITY contains dulaglutide, which is a human GLP-1 receptor agonist with 90% amino acid sequence homology to endogenous human GLP-1 (7-37). Dulaglutide activates the GLP-1 receptor, a membrane-bound cell-surface receptor coupled to adenylyl cyclase in pancreatic beta cells. Dulaglutide increases intracellular cyclic AMP (cAMP) in beta cells leading to glucose-dependent insulin release. Dulaglutide also decreases glucagon secretion and slows gastric emptying. INDICATIONS AND USAGE: 1.1 Limitations of Use HOW SUPPLIED: |
| These highlights do not include all the information needed to use BYETTA safely and effectively. See full prescribing information for BYETTA.
CLINICAL PHARMACOLOGY :Mechanism of Action: Incretins, such as glucagon-like peptide-1 (GLP-1), enhance glucose-dependent insulin secretion and exhibit other antihyperglycemic actions following their release into the circulation from the gut. BYETTA is a GLP-1 receptor agonist that enhances glucose-dependent insulin secretion by the pancreatic beta-cell, suppresses inappropriately elevated glucagon secretion, and slows gastric emptying. The amino acid sequence of exenatide partially overlaps that of human GLP-1. Exenatide has been shown to bind and activate the human GLP-1 receptor in vitro. This leads to an increase in both glucose-dependent synthesis of insulin, and in vivo secretion of insulin from pancreatic beta cells, by mechanisms involving cyclic AMP and/or other intracellular signaling pathways. Exenatide promotes insulin release from pancreatic beta cells in the presence of elevated glucose concentrations. BYETTA improves glycemic control by reducing fasting and postprandial glucose concentrations in patients with type 2 diabetes through the actions described below. Glucose-dependent insulin secretion: BYETTA has acute effects on pancreatic beta-cell responsiveness to glucose leading to insulin release predominantly in the presence of elevated glucose concentrations. This insulin secretion subsides as blood glucose concentrations decrease and approach euglycemia. However, BYETTA does not impair the normal glucagon response to hypoglycemia. First-phase insulin response: In healthy individuals, robust insulin secretion occurs during the first 10 minutes following intravenous (IV) glucose administration. This secretion, known as the "first-phase insulin response," is characteristically absent in patients with type 2 diabetes. The loss of the first-phase insulin response is an early beta-cell defect in type 2 diabetes. Administration of BYETTA at therapeutic plasma concentrations restored first-phase insulin response to an IV bolus of glucose in patients with type 2 diabetes (Figure 1). Both first-phase insulin secretion and second-phase insulin secretion were significantly increased in patients with type 2 diabetes treated with BYETTA compared with saline (p <0.001 for both). Pharmacokinetics: Distribution: Metabolism and Elimination: INDICATIONS AND USAGE: Important Limitations of Use The concurrent use of BYETTA with insulin has not been studied and cannot be recommended. Based on postmarketing data BYETTA has been associated with acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis. BYETTA has not been studied in patients with a history of pancreatitis. It is unknown whether patients with a history of pancreatitis are at increased risk for pancreatitis while using BYETTA. Other antidiabetic therapies should be considered in patients with a history of pancreatitis DOSAGE AND ADMINISTRATION: Use BYETTA only if it is clear, colorless and contains no particles. Renal Impairment: DOSAGE FORMS AND STRENGTHS: CONTRAINDICATIONS: WARNINGS AND PRECAUTIONS: ADVERSE REACTIONS: Postmarketing reports of increased international normalized ratio (INR) with concomitant use of warfarin, sometimes with bleeding. To report SUSPECTED ADVERSE REACTIONS contact Amylin Pharmaceuticals, Inc. and Eli Lilly and Company at 1-800-868-1190 and www.byetta.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONS: USE IN SPECIFIC POPULATIONS: |
| These highlights do not include all the information needed to use Victoza safely and effectively. See full prescribing information for Victoza.
Victoza® (liraglutide (rDNA origin) injection), solution for subcutaneous use -
INDICATIONS AND USAGE: Important Limitations of Use: DOSAGE AND ADMINISTRATION:
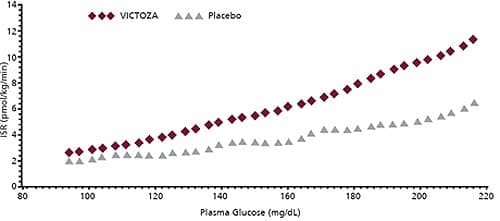
DOSAGE FORMS AND STRENGTHS: Solution for subcutaneous injection, pre-filled, multi-dose pen that delivers doses of 0.6 mg, 1.2 mg, CONTRAINDICATIONS: WARNINGS AND PRECAUTIONS: Pancreatitis: In clinical trials, there were more cases of pancreatitis among Victoza-treated patients than among comparator-treated patients. If pancreatitis is suspected, Victoza and other potentially suspect drugs should be discontinued. Victoza should not be restarted if pancreatitis is confirmed. Use with caution in patients with a history of pancreatitis. Serious hypoglycemia: Can occur when Victoza is used with an insulin secretagogue (e.g. a sulfonylurea). Consider lowering the dose of the insulin secretagogue to reduce the risk of hypoglycemia. Macrovascular outcomes: There have been no studies establishing conclusive evidence of macrovascular risk reduction with Victoza or any other antidiabetic drug. ADVERSE REACTIONS: To report SUSPECTED ADVERSE REACTIONS, contact Novo Nordisk Inc. at 1-877-484-2869 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS: USE IN SPECIFIC POPULATIONS: CLINICAL PHARMACOLOGY: GLP-1(7-37) has a half-life of 1.5-2 minutes due to degradation by the ubiquitous endogenous enzymes, dipeptidyl peptidase IV (DPP-IV) and neutral endopeptidases (NEP). Unlike native GLP-1, liraglutide is stable against metabolic degradation by both peptidases and has a plasma half-life of 13 hours after subcutaneous administration. The pharmacokinetic profile of liraglutide, which makes it suitable for once daily administration, is a result of self-association that delays absorption, plasma protein binding and stability against metabolic degradation by DPP-IV and NEP. Pharmacodynamics: Fasting and postprandial glucose was measured before and up to 5 hours after a standardized meal after treatment to steady state with 0.6, 1.2 and 1.8 mg Victoza or placebo. Compared to placebo, the postprandial plasma glucose AUC 0-300min was 35% lower after Victoza 1.2 mg and 38% lower after Victoza 1.8 mg. Glucose-dependent insulin secretion Figure 2: Mean Insulin Secretion Rate (ISR) versus Glucose Concentration Following Single-Dose Victoza 7.5 mcg/kg (~ 0.7 mg) or Placebo in Patients with Type 2 Diabetes (N=10) During Graded Glucose Infusion
Glucagon secretion: Gastric emptying: Cardiac Electrophysiology (QTc): Pharmacokinetics: Distribution - The mean apparent volume of distribution after subcutaneous administration of Victoza 0.6 mg is approximately 13 L. The mean volume of distribution after intravenous administration of Victoza is 0.07 L/kg. Liraglutide is extensively bound to plasma protein (>98%). Metabolism - During the initial 24 hours following administration of a single [3H]-liraglutide dose to healthy subjects, the major component in plasma was intact liraglutide. Liraglutide is endogenously metabolized in a similar manner to large proteins without a specific organ as a major route of elimination. Elimination - Following a [3H]-liraglutide dose, intact liraglutide was not detected in urine or feces. Only a minor part of the administered radioactivity was excreted as liraglutide-related metabolites in urine or feces (6% and 5%, respectively). The majority of urine and feces radioactivity was excreted during the first 6-8 days. The mean apparent clearance following subcutaneous administration of a single dose of liraglutide is approximately 1.2 L/h with an elimination half-life of approximately 13 hours, making Victoza suitable for once daily administration. Recommended Storage: After initial use of the Victoza pen, the pen can be stored for 30 days at controlled room temperature (59°F to 86°F; 15°C to 30°C) or in a refrigerator (36°F to 46°F; 2°C to 8°C). Keep the pen cap on when not in use. Victoza should be protected from excessive heat and sunlight. Always remove and safely discard the needle after each injection and store the Victoza pen without an injection needle attached. This will reduce the potential for contamination, infection, and leakage while also ensuring dosing accuracy. |
| Incretins are insulin secretagogues. The two main candidate molecules that fulfill criteria for being an incretin are glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (glucose-dependent insulinotropic peptide, GIP). Both GLP-1 and GIP are rapidly inactivated by the enzyme dipeptidyl peptidase-4 (DPP-4).
Glucagon-like peptide analogs and agonists Exenatide (also Exendin-4, marketed as Byetta) is the first GLP-1 agonist approved for the treatment of type 2 diabetes. Exenatide is not an analogue of GLP, but rather a GLP agonist. Exenatide has only 53% homology with GLP, which increases its resistance to degradation by DPP-4 and extends its half-life. Typical reductions in A1C values are 0.5-1.0%. Liraglutide, a once daily human analogue (97% homology), has been developed by Novo Nordisk under the brand name Victoza. Taspoglutide is presently in Phase III clinical trials with Hoffman-La Roche. These agents may also cause a decrease in gastric motility, responsible for the common side effect of nausea, and is probably the mechanism by which weight loss occurs." [source] |
| Drug UPDATES: Adlyxin™ (lixisenatide) injection [Drug information / PDF] Package insert - Dosing: Click (+) next to Dosage and Administration section (drug info link) Initial U.S. Approval: 2016 Mechanism of Action: INDICATIONS AND USAGE ADLYXIN is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
DOSAGE AND ADMINISTRATION HOW SUPPLIED:
|
National Institutes of Health, U.S. National Library of Medicine, DailyMed Database.
Provides access to the latest drug monographs submitted to the Food and Drug Administration (FDA). Please review the latest applicable package insert for additional information and possible updates. A local search option of this data can be found here.